

This post does a deep dive into the lesser understood of the two known factors that cause Alzheimer’s Disease (AD) to progress and degrade brain function. It’s called Tau protein and until recently hasn’t gotten nearly as much attention as its sibling amyloid-beta protein. After making my head spin for weeks, I’ll admit this is a lot to digest, but hopefully by me sharing how I’ve made sense of it as a layperson makes it easier for you as well. If you have more down-to-earth insight, or corrections to how I’ve understood this highly technical medical specialty, please share as well in the comments so we can all understand better!
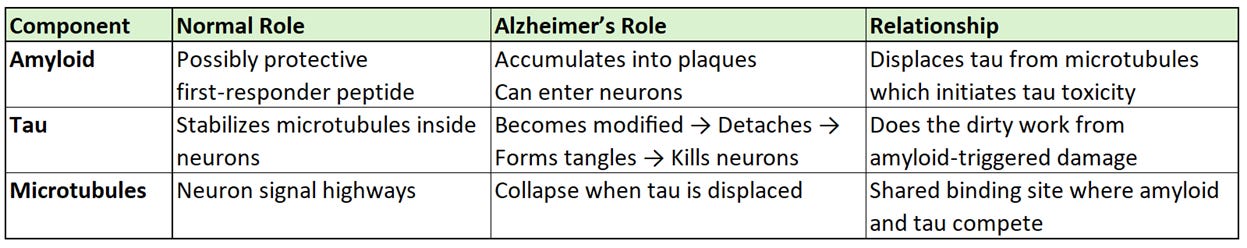
Tau is a microtubule‑associated protein found primarily inside neurons in the brain. Its normal job is to stabilize microtubules, which are the internal highways that transport nutrients and signals within neurons. Tau binds and unbinds to tubules dynamically, helping microtubules grow, shrink, and adapt to the cell’s needs.
When tau is functioning normally, neurons maintain their structure and communication pathways. However, in AD, tau undergoes harmful changes:
Tau becomes chemically modified or “phosphorylated”. Phosphorylated tau (p-tau) is tau protein that has had phosphate groups added to it. This occurrence is normal in small amounts, but in AD it becomes hyperphosphorylated.
Once hyperphosphorylated, the modified tau detaches from microtubules, causing the microtubule network to collapse and damaging the neurons.
Detached tau aggregates into neurofibrillary tangles, which form inside neurons.
These tangles are toxic and strongly correlate with neurodegeneration and cognitive decline.
In fact, the number and location of tau tangles in the brain track more closely with memory loss than amyloid plaque accumulation does, but they both perform a dance together that results in the devastation we know as Alzheimer’s Disease. This relationship, or dance, is where the knowledge and research has shifted dramatically in the last few years.
Recent studies show that Amyloid can bind to microtubules with similar strength as tau, and when Amyloid accumulates inside neurons, it can displace tau from microtubules. This displacement may be the initial trigger for tau dysfunction and tangle formation.
This amyloid-tau dance unifies decades of conflicting amyloid‑centric and tau‑centric theories into a new understanding that amyloid acts upstream of tau.
Studies show that amyloid exposure accelerates tau tangle formation, and amyloid-induced synaptic dysfunction and neuron death are tau‑dependent. That means that amyloid toxicity requires tau to do the real damage of AD.
Think of it this way … amyloid is the trigger, and tau is the bullet that actually kills neurons.
It gets worse… clinical evidence suggests that amyloid drives tau into a toxic state, and then toxic tau can further enhance amyloid toxicity, creating a self‑reinforcing dance of death.
Some new studies also show that certain groups (e.g., Black and Hispanic adults) show higher tau levels even before amyloid buildup. This suggests that additional biological or social factors can also influence tau pathology further clarifying how complicated AD is and that its causes, progression, and biomarkers are not identical across different populations in our society.
So, you’ve now heard about three key components related to neuronal damage from Alzheimer’s disease and I’ve summarized their normal and beneficial role in the brain, their toxic role in AD, and their relationship to one another in the table below:
Scientists’ evolving understanding of the disease is reshaping therapeutic strategies too and three areas of focus have developed:
Amyloid‑clearing therapies (e.g., lecanemab & donanemab) aim to reduce the upstream trigger.
Tau‑targeting therapies (antisense oligonucleotides, vaccines, aggregation inhibitors) aim to block the downstream damage.
Microtubule‑stabilizing approaches (e.g., lithium) may protect the shared binding site.
The future likely involves cocktail-like treatment combinations, or stage‑specific therapy, guided by biomarkers like Amyloid-beta42/40, p‑tau217, and tau PET.
Though I went over several anti-amyloid treatments in my prior article, the comparison below shows how the current, as well as a few emerging, anti-amyloid treatments effect tau levels in the body.
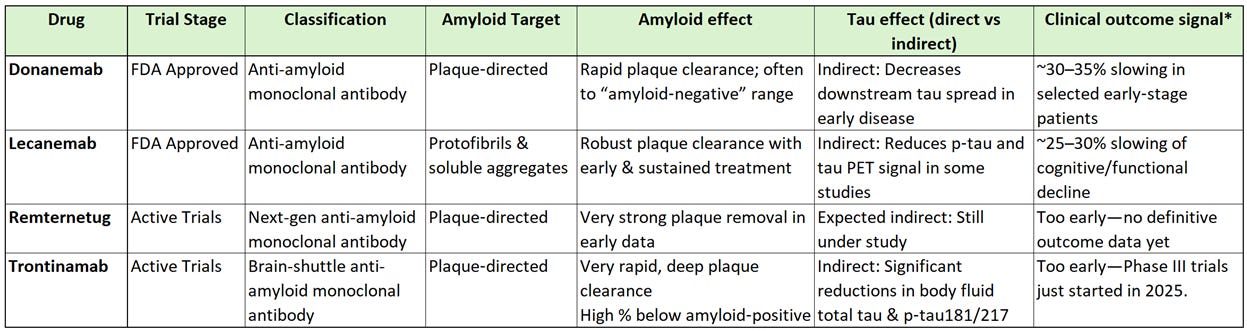
*Clinical outcome signal is the effect on composite cognitive/functional scales in early symptomatic AD
All currently approved AD treatments are amyloid‑lowering monoclonal antibodies. None of them are tau‑directed therapies, but tau biomarkers still move. When amyloid is lowered early enough and deeply enough, several trials show reduced p-tau levels in cerebrospinal fluid (CSF) & blood plasma (e.g., p‑tau181, p‑tau217), as well as slower increase in tau PET signal in early‑stage patients.
In human-speak… remove the trigger (amyloid-beta plaque), and you dampen the rate at which tau pathology spreads. Keep in mind these don’t erase existing tangles because you can’t undo existing neuronal damage.
Emerging Tau Treatment Therapies
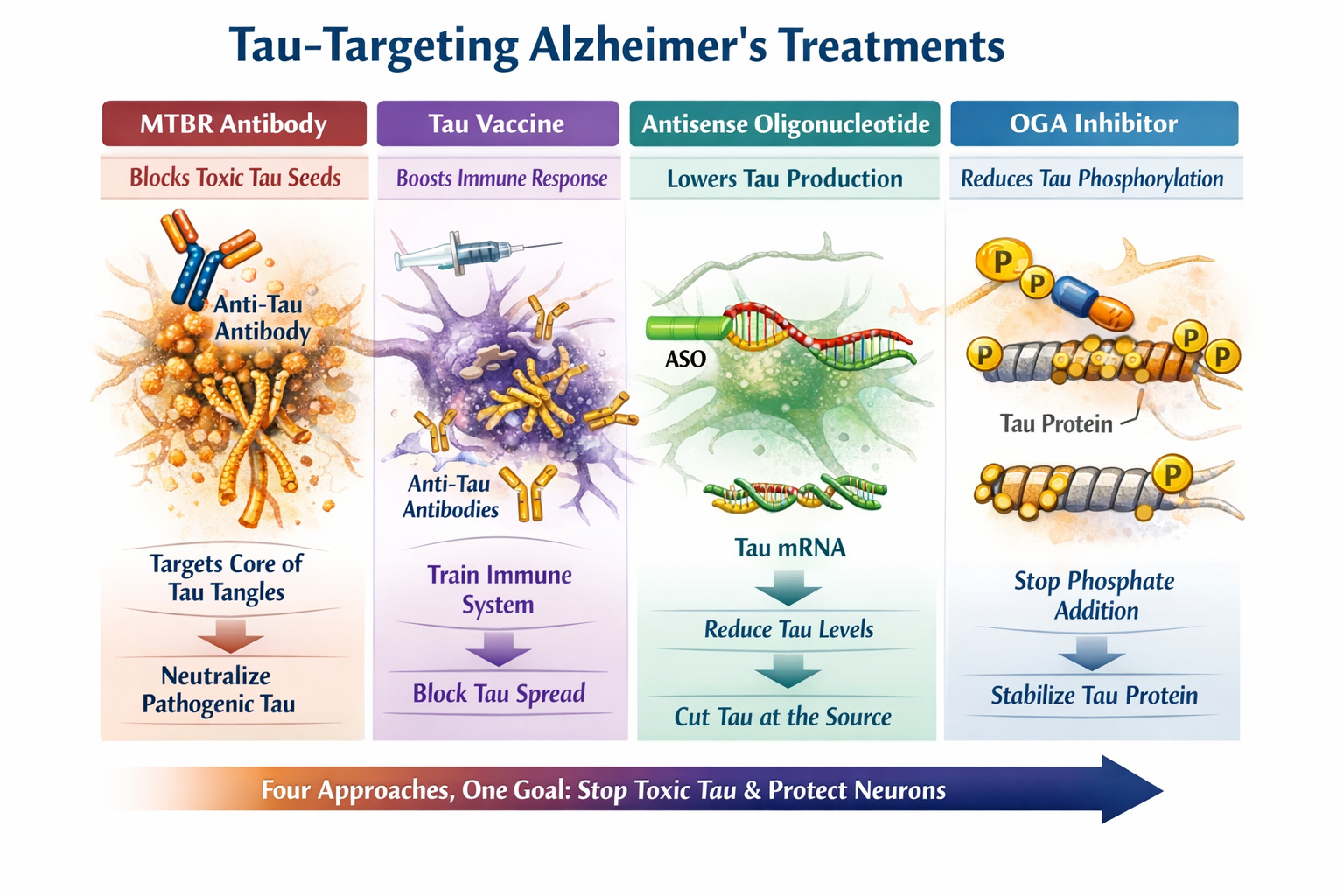
Though tau research is significantly behind that of amyloid, there are multiple tau‑targeting therapies in clinical trials right now. They include novel approaches like monoclonal antibodies, antisense oligonucleotides, kinase inhibitors, aggregation blockers, and O‑GlcNAcase inhibitors. Several are already in Phase II and Phase II/III.
Passive Immunotherapies (Tau Monoclonal Antibodies) aim to bind to extracellular tau seeds and block neuron‑to‑neuron spread.
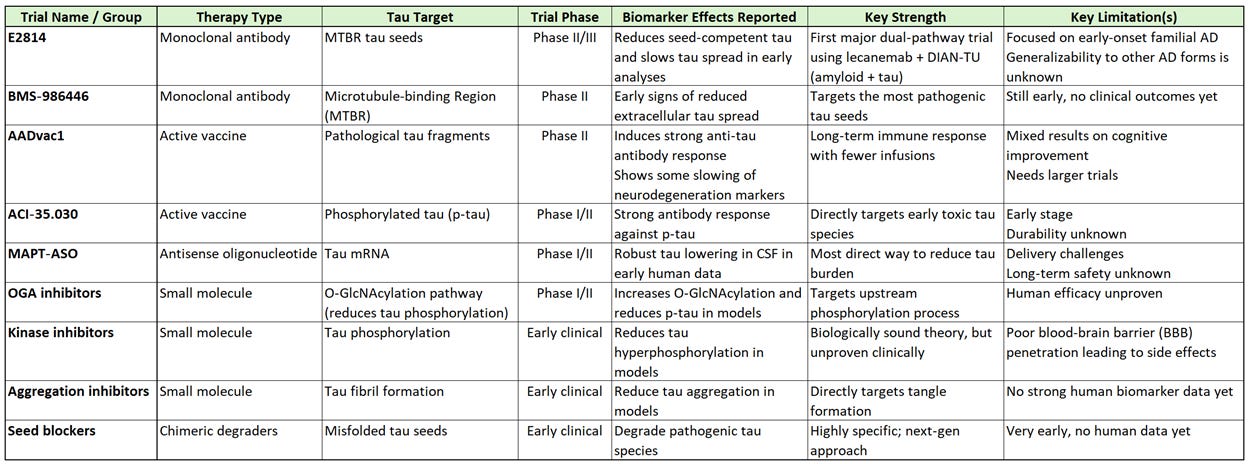
BMS‑986446 (TargetTau‑1 trial) binds the microtubule binding region (MTBR), which is the core of pathogenic tau, to prevent inter-neuronal transmission of tau aggregates. MTBR‑targeting is considered more promising than earlier antibodies that failed.
E2814 (DIAN‑TU platform trial) MTBR‑targeting antibody designed to neutralize tau seeds and is combined with lecanemab as the first major trial testing amyloid removal + tau removal together in early-onset familial (also known as autosomal‑dominant) AD.
Active Immunotherapies (Tau Vaccines) stimulate the immune system to produce anti‑tau antibodies.
AADvac1 targets pathological tau fragments
ACI‑35.030 targets phosphorylated tau with the goal of long‑term suppression of tau spread with fewer infusions.
Antisense Oligonucleotides (ASO) reduce tau production at the RNA level.
MAPT‑ASO programs lower total tau in neurons. Humans tolerate partial tau reduction well, making this a viable strategy.
Sounds scary to me to say that “Humans tolerate x well…” I’m sure someone back in the 1970s said, “Humans tolerate ingesting the chemicals that leach out of plastic well, making plastic food containers a viable strategy.”
Tau Aggregation Inhibitors aim to prevent tau from forming fibrils or break up existing aggregates.
Small molecules that block MTBR‑mediated aggregation
Seed‑disrupting compounds targeting early oligomers
Post‑Translational Modification Modulators - Tau becomes toxic through phosphorylation, acetylation, glycosylation, and cleavage. Several drug classes target these steps.
O‑GlcNAcase (OGA) inhibitors increase O‑GlcNAcylation, which prevents tau phosphorylation and aggregation.
Kinase inhibitors target kinases which are enzymes that add phosphate to another molecule and, in turn, drives tau hyperphosphorylation in the brain.
Acetylation modulators aim to reverse tau acetylation. When proteins like Tau are chemically modified with an acetyl group, they become acetylated, are harder to clear, and begin to aggregate or clump.
Misfolded Tau Seed Blocker Therapies target tau seeds directly. Tau “seeds” are small, highly pathogenic aggregates that initiate disease spread.
Includes DEPTAC, PhosTAC, and other chimeric molecules. Chimeric molecules are hybridized molecules that fuse parts of different molecules together. Part A performs one task and Part B performs another so they function unlike any other known molecule. It has huge promise in AD treatment where it may be able to recognize rogue proteins (part A), isolate them (part B), and remove them (part C).
Oy that’s a lot to unpack! Here’s a simpler graphic depiction I created of the investigative treatment areas… (TBT I’ve gotten pretty good at AI client prompts and many of these graphics, as well as my article cover art, are AI-generated, as is this one)
Tau‑targeting Treatment Comparison
To make all that info on clinical trials a little easier to digest for you data junkies like me, I created a comparison table. As you can see, we are likely years away from any of these making it to an FDA-approved treatment. Most are either highly specialized, or concepts that will more likely yield real human benefits a few revisions from now.
I don’t see these as something that will help someone like me with the clock already ticking, but I must admit some are very exciting! The whole field of chimeric molecules seems cutting edge but far off… chimeric compounds also occur in nature where genes have been know to accidentally fuse and viruses have been known to swap segments to form new hybrids. Chimeric molecules can occur because of mutation, recombination, or evolutionary pressure. Now scientists are using that same process to potentially cure diseases. Did you know that they’re called “chimeric” after the chimera in Greek mythology, which is a creature made of parts from 3 different animals… the head of a lion, the body of goat, and the tail of a serpent?

Does your brain hurt after reading all that? Mine sure did after researching, summarizing, simplifying, and sharing it! It felt like an endless spiral at times to convert medical gibberish into something that felt even remotely close to English. I’m sure other people have felt the same at times with my dissertations on emerging compute or communications technologies in the past!
In any case, I hope you came away feeling like you understand a bit more about amyloid and tau proteins and their relationship to Alzheimer’s disease now. I plan to release one more article on these proteins that nets it out from my perspective.
Coming Next: Month Seven Personal Update
Peace my Friends and Thanks for Reading!

Note: What I’m sharing in my deep dive newsletter articles is based on personal research, but please remember I am not in the healthcare profession, and I have no credentials in that area past or present. I’m sharing what I’ve learned, and will continue to learn, in my own desperate effort to advocate for myself when meeting with my care team. I believe everything I share to be correct or I wouldn’t share it, but you should not take anything I say as medical advice.